
The Korle Bu Teaching Hospital's Child Health Department recently documented a rare occurrence of Wilson's disease.
A 13-year-old boy has been affected by the disease.
This case marks the first of its kind in the country, with statistics indicating it typically affects one individual per 30,000 in developed nations.
The diagnosis came to light when the boy's mother, a trader, noticed a decline in his handwriting, prompting medical attention.
Professor Ebenezer Badoe, Head of the Neuro-Developmental Clinic within the Department of Child Health at KBTH, conducted thorough examinations leading to the diagnosis.
Despite the severity of the genetic disorder, Professor Badoe conveyed optimism regarding treatment possibilities.
While the condition is treatable with medication, he expressed concern over the financial burden it poses.
Sustaining the boy's health would require approximately GH¢450 per month, emphasizing the necessity for ongoing support to ensure his well-being.
All you need to know about Wilson disease
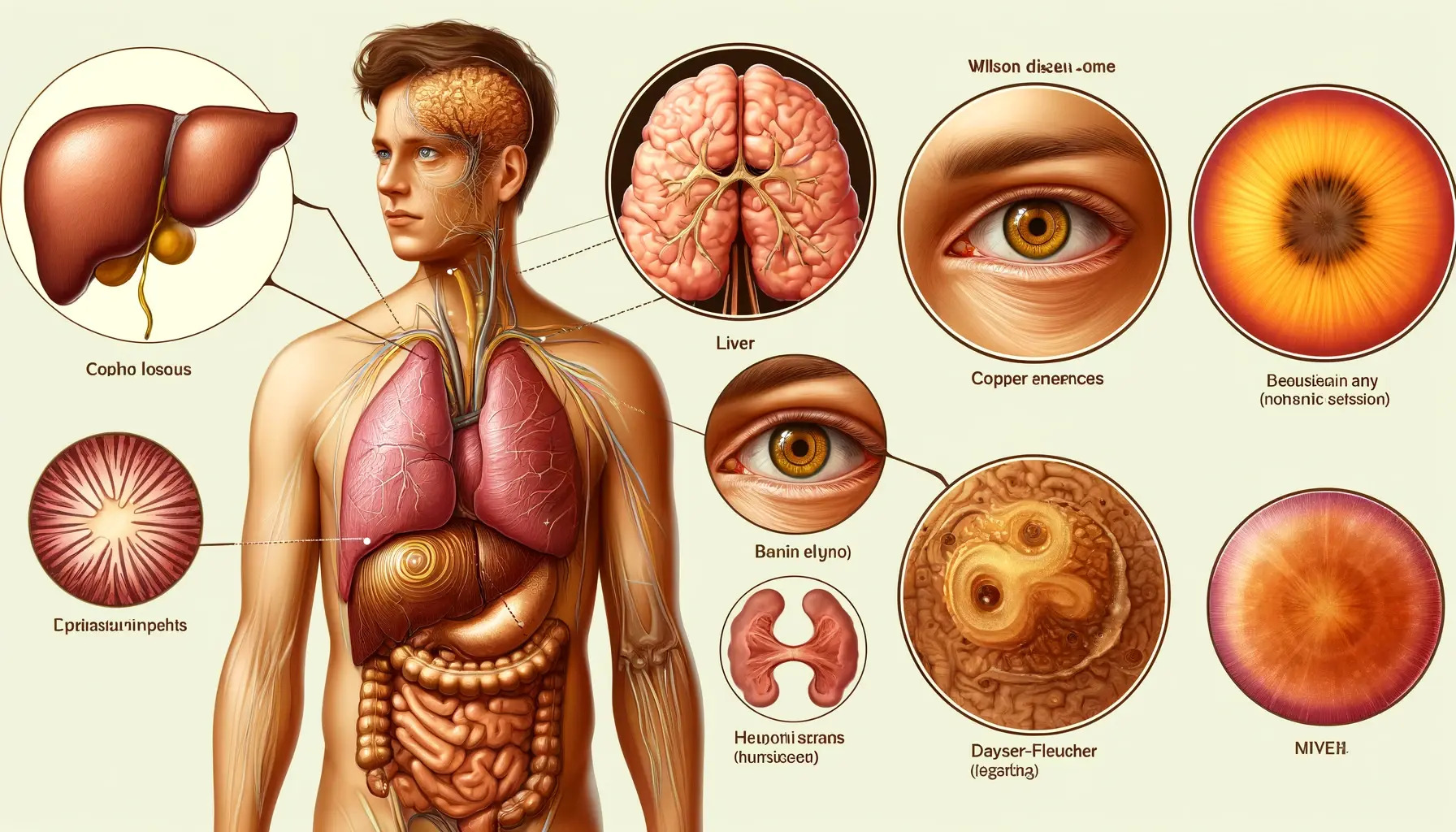
Wilson disease, also known as hepatolenticular degeneration, is a rare inherited disorder that leads to excessive accumulation of copper in the body. Here's a comprehensive overview:
Cause
Wilson disease is caused by mutations in the ATP7B gene, which encodes a protein responsible for regulating copper transport and elimination. These mutations impair the body's ability to excrete copper, leading to its accumulation, primarily in the liver and brain.
Symptoms
Symptoms can vary widely and may affect multiple organ systems:
- Hepatic (Liver) symptoms:
- Hepatitis
- Liver failure
- Cirrhosis
- Jaundice
- Abdominal pain
Neurological symptoms:
- Tremors
- Poor coordination
- Difficulty speaking
- Dystonia (muscle contractions)
- Behavioral changes
Psychiatric symptoms:
- Depression
- Anxiety
- Psychosis
- Personality changes
Other Symptoms:
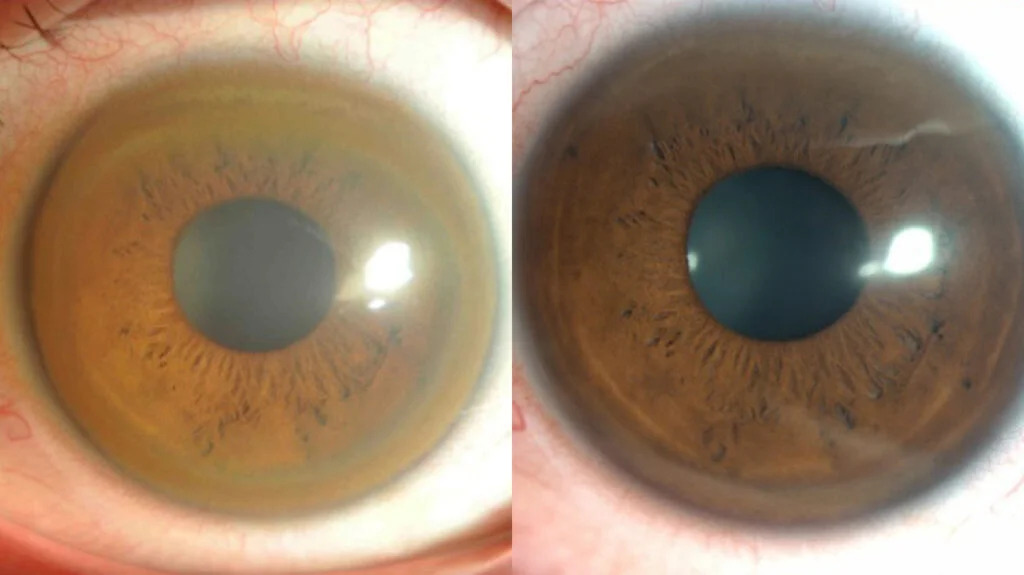
- Kayser-Fleischer rings (copper deposits in the cornea)
- Hemolytic anemia
- Diagnosis
Diagnosing Wilson disease typically involves a combination of the following:
Clinical evaluation: Physical examination for signs such as Kayser-Fleischer rings.
Laboratory tests:
Serum ceruloplasmin levels (often low in Wilson disease)
Serum copper levels (can be low, but free copper is elevated)
24-hour urinary copper excretion (usually elevated)
Liver Biopsy: To measure hepatic copper content.
Genetic Testing: Identifying mutations in the ATP7B gene.
Treatment
The main goal of treatment is to reduce copper accumulation and prevent its effects. Treatments include:
Chelation therapy:
- Penicillamine or Trientine: Medications that bind to copper and help its excretion through urine.
- Zinc Acetate: Increases copper excretion via stool by blocking copper absorption in the intestines.
Dietary modifications:
Avoiding copper-rich foods (e.g., shellfish, liver, mushrooms, nuts, chocolate).
Liver Transplant: In severe cases, particularly those with advanced liver disease or acute liver failure.
Prognosis
With early diagnosis and appropriate treatment, many individuals with Wilson disease can lead normal lives.
However, untreated Wilson disease can be fatal due to liver failure or severe neurological impairment.
Regular follow-up is crucial for managing Wilson disease. This includes monitoring copper levels, liver function tests, and adherence to treatment protocols.
Ongoing research aims to better understand Wilson disease and develop more effective treatments. Gene therapy and new chelating agents are areas of active investigation.
Read Full Story




















Facebook
Twitter
Pinterest
Instagram
Google+
YouTube
LinkedIn
RSS